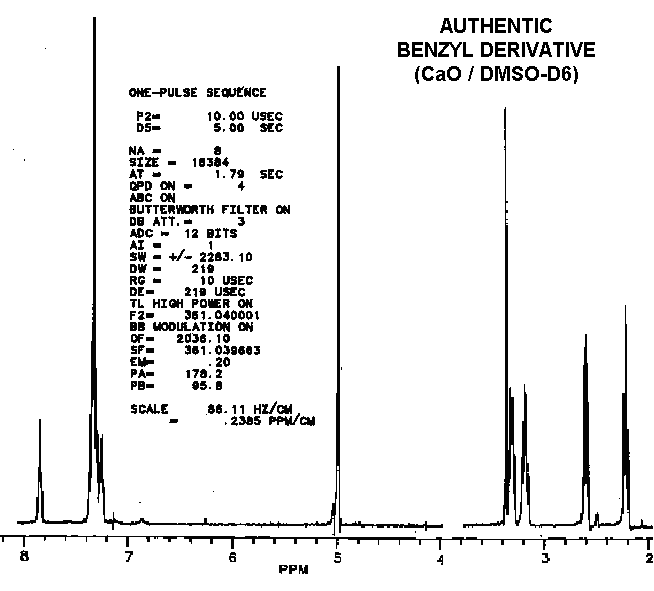
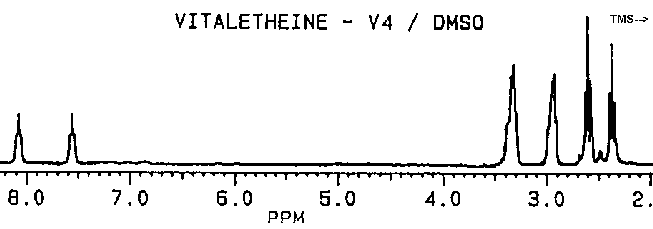
As observed by others, the "benzyl derivative" produces a different spectra in water than in DMSO. This indicates a potential for dehydrations which ironically might be water-catalyzed. Subtle differences in the proton NMR spectra (especially in the hydrogen-bonded moieties) between authentic vitaletheine V4 and other's preparations suggest rearrangements and decompositions, including decarboxylations, that also may be dependent upon DMSO, water, and hypobaric conditions.
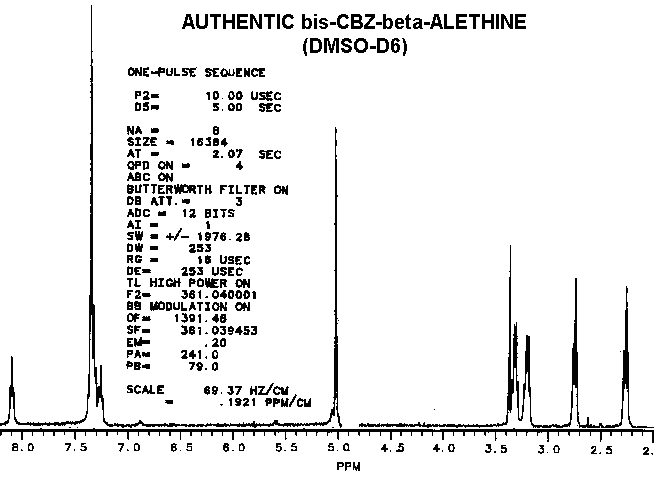
Considering that the benzyl derivative is soluble in water and the CBZ-blocked starting material is not, these two compounds have remarkably similar proton NMR spectra.
The technical expertise of Dr. Cary J. Morrow, then chairman of the
Department of Chemistry at UNM, in obtaining the following spectra is gratefully
acknowledged.
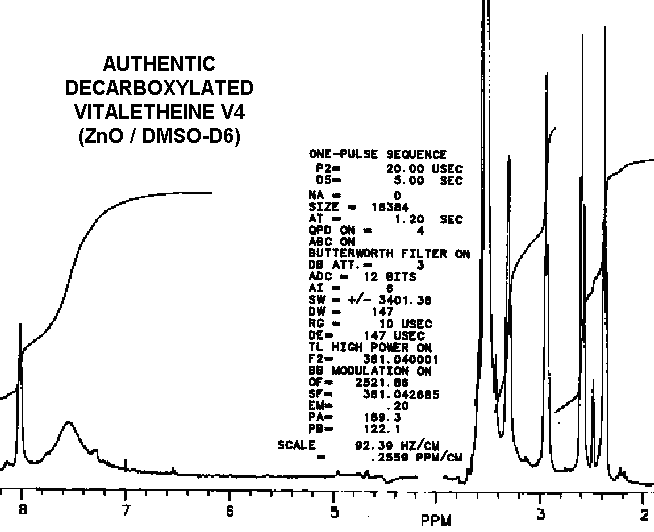
Removal of the carbobenzoxyl moiety, such as in decarboxylated vitaletheine V4, produces some dramatic changes in both coupling and in the relative intensities of the peaks in the proton spectra for the resulting free amine; the integrated signal associated with the proton in the carbamate moiety nearly triples upon decarboxylation. Removal of the carboxyl moiety also causes changes in the coupling of the adjacent methylene at about 3 ppm (from a quartet to a broadened triplet) indicating coupled amino protons that are more rapidly exchangeable than those in the carbamate. The formation of a carbonimidic tautomer should have a similar effect upon this methylene's coupling, but an increase in protonation of the adjacent amine cannot be explained by this structure since the carbonimidic tautomer by definition has protons associated only with its oxygens and not with the nitrogen.
The fact that decarboxylation of vitaletheine V4 occurring in DMSO does
not radically affect the positions of the methylenes in its spectra suggests
that DMSO causes structural changes in vitaletheine V4 prior to decarboxylation,
possibly by removing zinc ions and thereby destabilizing the zinc complex.
Compared to aqueous solutions, dissolving the benzyl derivative in DMSO
also causes an
upfield shift in the methylenes
thought to be coupled with the sulfur moieties. Since this solvent-dependent
upfield shift is even more obvious with vitaletheine V4, removal of a deshielding
influence (i.e., the stabilizing cation) by DMSO seems likely.
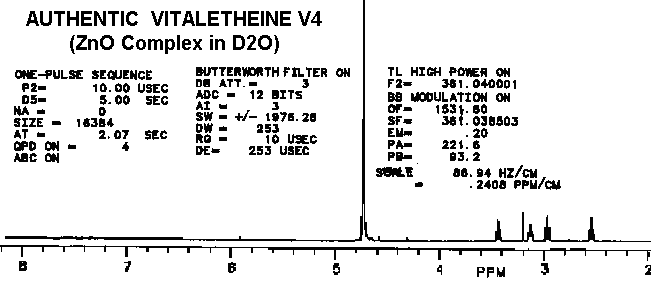
The proton spectra of the product from partially-reduced benzyl derivative
is nearly identical with vitaletheine V4, possibly helping to explain some
of the
biological activities of the benzyl
derivative.
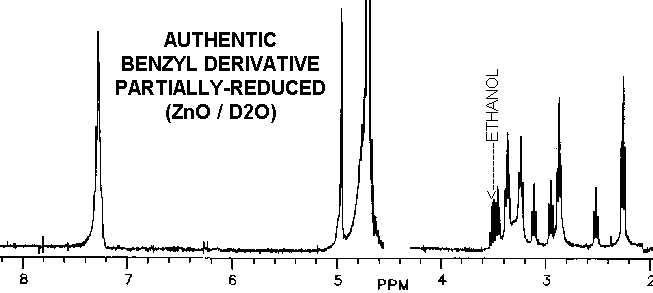
The reduced benzyl derivative and vitaletheine V4, though similar, are
clearly distinct from ß-aletheine prepared in a similar fashion.
Also, since decarboxylated vitaletheine V4 differs from ß-aletheine
only by its polymerization through the carbonyls of its amides, the spectra
of these two compounds are very similar, yet distinct.
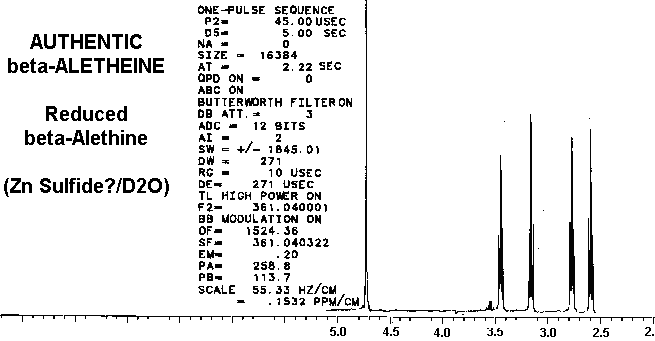
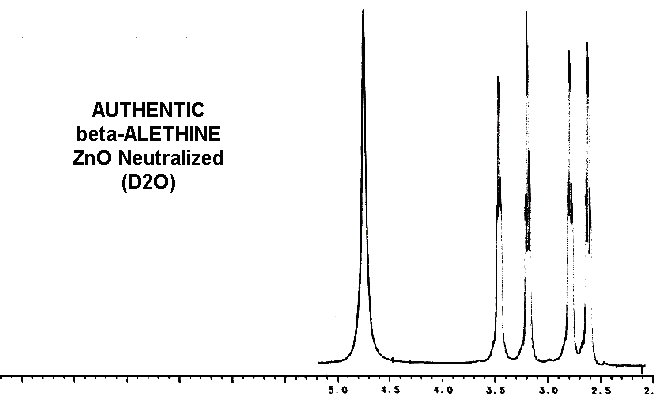
Considering these similarities in the spectra of reduced benzyl derivative,
ß-aletheine, and vitaletheine V4, there is very little spectral evidence
in these authentic preparations for
the higher
oxidation states alleged by others.
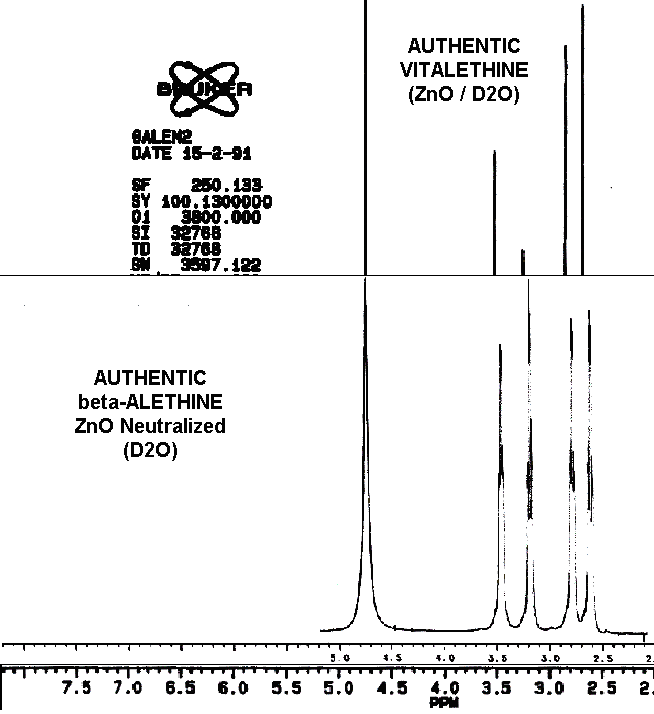
The spectral differences between ß-alethine and vitalethine are
nearly imperceptible. The pattern of peak intensities is the most obvious
of these differences, as the following overlay illustrates.
GO TO:
| Home | Overview | People | Journal | Nutrition |
| Environment |
|
WWW Links | Outline | e-mail us |
{kind=link}