Copyright
©
1996, 1997, 1999, 2000, 2001, 2002, 2003, 2006, 2008 by Galen D. Knight
and
Vitaletherapeutics, Inc.
CURRICULUM VITAE
Galen Daryl Knight, Ph.D.
3205 Arizona, NE, Albuquerque, New Mexico, USA
87110-2640
e-mail: galenvtp@highfiber.com
Voice: (505) 884-8644 Fax:(505) 884-1388
I. QUALIFICATIONS AND BACKGROUND:
A. SUMMARY OF GALEN'S WORK EXPERIENCE AND ACCOMPLISHMENTS:
Galen has been doing health research in various areas of interest for more than 30 years, having spent large portions of his career i) identifying the true iodinating system in the thyroid that probably produces thyroid hormone and ii) elucidating new ways in which cholesterol biosynthesis is regulated, and iii) elucidating what causes cancer and non-toxic ways of preventing and treating it. Cancer and heart disease (caused for the most part by very similar nutritional and immunological deficiencies, and toxic environmental factors) kill 68% of the Americans that die every year. Thyroid imbalances, also resulting from perturbations in these same factors, can result in a variety of health problems, including poor assimilation and utilization of food, poor thermal regulation, imbalances in enzymes and protein, and even neurological and "emotional" problems such as delirium, schizophrenia, hallucinations, mania, seizures, and anxiety.
Galen may be the only person who has chemically synthesized all of the authentic, exemplary compounds for the beta-alethine and vitaletheine modulator technologies. Consequently, he was and is intrinsically involved in i) theories developed before his employment at UNM ii) creating new theories and approaches to the treatment of disease, iii) identifying and purifying novel biologically active compounds, iv) working out the protocols for chemically synthesizing them, v) making them, vi) definitively establishing their chemical structures, vii) identifying appropriate biological assays for studying these types of compounds, and viii) testing the biological activities of these synthetic compounds in a variety of tumor, cell culture, and cell-free (test tube) biological assays.
As part of his efforts to help develop these technologies, Galen i) conducted most of the prior art searches in the chemical literature and in much of the patent literature before any patent applications were filed, ii) drafted large portions of the patent applications, and iii) formulated most of the science behind the generic structures for the original patent applications, such as that resulting in US 5,370,868. He also drafted scientific manuscripts that were later published in back-to-back papers in the November 1, 1994, issue of the prestigious scientific journal, Cancer Research, and responded to office actions for filed patent applications when requested.
Galen has been listed as inventor on more than a hundred patents and applications, world-wide, on the beta-alethine and vitaletheine modulator technologies and is the author of most of a web site devoted to the study and dissemination of information about these and other inexpensive, non-toxic, nutritional, environmental, and immunotherapeutic approaches to the treatment and elimination of disease.
SELECTED PUBLICATIONS
- VitaleTherapeutics, Inc.'s Web Site (copyright © 1996, 1997, 1998, 1999, 2000, 2001, 2002, 2003, 2004, 2005, 2006),
- Knight, G. D. In: Pitfalls in Synthesizing Vitaletheine Modulators and associated links.
- Ramos, S. M., O'Donnell, L. S., Knight, G. 1999. Edema volume, not timing, is the key to success in lymphedema treatment. Am. J. Surg., 178: 311-315.
- Knight, G. D., Laubscher, K. H., Fore, M. L., Clark, D. A., and Scallen, T. J. 1994. Vitalethine modulates erythropoiesis and neoplasia. Cancer Res., 54: 5623-5635.
- Knight, G. D., Mann, P. L., Laubscher, K. H., and Scallen, T. J. 1994. Seemingly diverse activities of beta-alethine. Cancer Res., 54: 5636-5642.
- Johnson, K. A., Morrow, C. J., Knight, G. D., and Scallen, T. J. 1994. In vivo formation of 25-hydroxycholesterol from endogenous cholesterol after a single meal, dietary cholesterol challenge. J. Lipid. Res., 35(12): 2241-2253.
- Knight, G. D., and Scallen, T. J. Use of vitaletheine modulators in the prophylaxis and treatment of disease. US Patent Application.
-
Knight, G. D. Resolution and reconstitution of the NADPH-dependent tyrosyl- peptide iodinating activity from porcine thyroid tissue. In: Dissertation. Austin, Texas: The University of Texas at Austin, 1982. See note added in proof: http://www.highfiber.com/~galenvtp/ipreface.htm
The
significance of these works is that they establish a novel mechanism
for control of virtually every metabolic pathway in the body. Most
proteins,
peptides, and enzymes i) have L-cysteine residues that stabilize, while
also ii) regulating the metabolism of these substances through
interactions with
small-molecular-weight sulfur-containing compounds thereby influencing
their
biological activities. Such insights enable a different approach to the
treatment of disease, one that capitalizes upon the subtle effects of
modulating
a number of nutritional and other factors that together, through their
synergism,
cause a greatly enhanced response from the various amplifying enzyme
cascades within our bodies. The hydroxycholesterol work is highly
significant,
because this artifact occurs in processed food products, probably via
oxidation
through a 24,25-epoxide, that is inherently toxic to thiol compounds
such
as vitaletheine and that have been shown to
poison
cancer-fighting immune responses. The identification of other environmental factors that poison these
regulatory
pathways establishes an integrative approach for identifying
environmental
and nutritional causes of disease and of new ways for intervening
metabolically
and immunologically to address these underlying causes.
PATENTS
The full extent of patents and applications,
listing Knight as inventor, is not known at this time due to i) changes
in coverage due to prosecution, ii) rejection, iii) abandonment, and
iv) issuance of new patents, and v)
pending Federal Circuit resolution of other issues. The current status
with regards to issued patents can be obtained from various patent
databases.
Included below is a partial list that was obtained at one time from a
name
search of "Knight Galen" on one such database, excluding patents known
to contain information erroneously modified by others. Discovery
obtained in the federal lawsuit and appeals all the way to the US
supreme court has demonstrated both, fraudulent modifications of data
from Galen's and Scallen's work and a willingness of the USPTO, the
federal court system, the University of New Mexico, and its now defunct
licensee led by Floyd Taub and his accomplices, to cover up and
continue to maintain this corruption of data.
|
Knight, Galen D. |
Mann, Paul L. |
Scallen, Terence J. |
|
PATENT #S |
ISSUED |
TITLE |
|
WO9200960A1 |
Jan. 23/1992 |
Beta-Alethine Use in Cell Culture and Therapy |
|
US6046241 |
April 04/2000 |
ß-alethine as Anti-Tumor Agent |
|
US5643966 |
July 01/1997 |
ß-alethine as Anti-Tumor Agent |
|
SU9200960W1 |
Jan. 23/1992 |
|
|
SE538330R4 |
Oct. 11/1995 |
|
|
SE538330R1 |
April 28/1993 |
|
|
SD9200960W1 |
Jan. 23/1992 |
|
|
RO9200960W1 |
Jan. 23/1992 |
|
|
PL9200960W1 |
Jan. 23/1992 |
|
|
OA9200960W1 |
Jan. 23/1992 |
|
|
NO9200960W1 |
Jan. 23/1992 |
|
|
NL538330R4 |
Oct. 11/1995 |
|
|
NL538330R1 |
April 28/1993 |
|
|
MW9200960W1 |
Jan. 23/1992 |
|
|
MG9200960W1 |
Jan. 23/1992 |
|
|
MC9200960W1 |
Jan. 23/1992 |
|
|
LU538330R4 |
Oct. 11/1995 |
|
|
LU538330R1 |
April 28/1993 |
|
|
LK9200960W1 |
Jan. 23/1992 |
|
|
LI538330R4 |
Oct. 11/1995 |
|
|
LI538330R1 |
April 28, 1993 |
|
|
KR9200960W1 |
Jan. 23/1992 |
|
|
KP9200960W1 |
Jan. 23/1992 |
|
|
JP9200960W1 |
Jan. 23/1992 |
|
|
JP5508769T2 |
Dec. 09/1993 |
|
|
IT538330R4 |
Oct. 11/1995 |
|
|
IT538330R1 |
April 28/1993 |
|
|
HU9200960W1 |
Jan. 23/1992 |
|
|
GR3018632T3 |
April 30/1996 |
Beta-Alethine Use in Cell Culture and Therapy |
|
GR538330R4 |
Oct. 11/1995 |
|
|
GR538330R1 |
April 28/1993 |
|
|
GB538330R4 |
Oct. 11/1995 |
|
|
GB538330R1 |
April 28/1993 |
|
|
FR538330R4 |
Oct. 11/1995 |
|
|
FR538330R1 |
April 28/1993 |
|
|
FI9200960W1 |
Jan. 23/1992 |
|
|
ES2080954T3 |
Feb. 16/1996 |
Uso de Beta-Aletina en Cultivos de Celulas y en Terapia |
|
ES538330R4 |
Oct. 11/1995 |
|
|
ES538330R1 |
April 28/1993 |
|
|
EP9200960W1 |
Jan. 23/1992 |
|
|
EP538330B1 |
Oct. 11/1995 |
Beta-Alethine Use in Cell Culture and Therapy |
|
EP538330A4 |
July 07/1993 |
Beta-Alethine Use in Cell Culture and Therapy |
|
EP538330A1 |
April 28/1993 |
Beta-Alethine Use in Cell Culture and Therapy |
|
DK9200960W1 |
Jan. 23/1992 |
|
|
DK538330T3 |
Feb. 26/1996 |
Beta-Alethin-Anvendelse til Celledyrkning og Terapi |
|
DK538330R4 |
Oct. 11/1995 |
|
|
DK538330R1 |
April 28/1993 |
|
|
DE69113802T2 |
May 15/1996 |
Anwendung von Beta-Alethin in Zellkultur und Therapie |
|
DE69113802C0 |
Nov. 16/1995 |
Anwendung von Beta-Alethin in Zellkultur und Therapie |
|
DE538330R4 |
Oct. 11/1995 |
|
|
DE538330R1 |
April 28/1993 |
|
|
CH538330R4 |
Oct. 11/1995 |
|
|
CH538330R1 |
April 28/1993 |
|
|
CA2087883AA |
Jan. 28, 1994 |
Beta-Alethine Use in Cell Culture and Therapy |
|
BR9200960W1 |
Jan. 23/1992 |
|
|
BG9200960W1 |
Jan. 23/1992 |
|
|
BE538330R4 |
Oct. 11/1995 |
|
|
BE538330R1 |
April 28/1993 |
|
|
BB9200960W1 |
Jan. 23/1992 |
|
|
AU9200960W1 |
Jan. 23/1992 |
|
|
AU8207791A1 |
Feb. 04/1992 |
Beta-Alethine Use in Cell Culture and Therapy |
|
AU646875B2 |
March 10/1994 |
Beta-Alethine Use in Cell Culture and Therapy |
|
AT538330R4 |
Oct. 11/1995 |
|
|
AT538330R1 |
April 28/1993 |
|
|
AT128964E |
Oct. 15/1995 |
Anwendung von Beta-Alethin in Zellkultur und Therapie |
|
Inventors: |
Knight, Galen D. |
Scallen, Terence J. |
|
PATENT #s |
ISSUED |
TITLE |
|
WO9200955A1 |
Jan. 23/1992 |
Vitaletheine and Use in Cell Culture and Therapy |
|
US5370868 |
Dec. 06/1994 |
Therapeutic use of vitaletheine modulators in neoplasia |
|
SU9200955W1 |
Jan. 23/1992 |
|
|
SE539525R4 |
Oct. 27/1999 |
|
|
SE539525R1 |
May 05/1993 |
|
|
SD9200955W1 |
Jan. 23/1992 |
|
|
RO9200955W1 |
Jan. 23/1992 |
|
|
PL9200955W1 |
Jan. 23/1992 |
|
|
OA9200955W1 |
Jan. 23/1992 |
|
|
NO9200955W1 |
Jan. 23/1992 |
|
|
NL539525R4 |
Oct. 27/1999 |
|
|
NL539525R1 |
May 05/1993 |
|
|
MW9200955W1 |
Jan. 23/1992 |
|
|
MG9200955W1 |
Jan. 23/1992 |
|
|
MC9200955W1 |
Jan. 23/1992 |
|
|
LU539525R4 |
Oct. 27/1999 |
|
|
LU539525R1 |
May 05/1993 |
|
|
LK9200955W1 |
Jan. 23/1992 |
|
|
LI539525R4 |
Oct. 27/1999 |
|
|
LI539525R1 |
May 05/1993 |
|
|
KR9200955W1 |
Jan. 23/1992 |
|
|
KP9200955W1 |
Jan. 23/1992 |
|
|
JP9200955W1 |
Jan. 23/1992 |
|
|
JP6500106T2 |
Jan. 6/1994 |
|
|
IT539525R4 |
Oct. 27/1999 |
|
|
IT539525R1 |
May 05/1993 |
|
|
HU9200955W1 |
Jan. 23/1992 |
|
|
GR539525R4 |
Oct. 27/1999 |
|
|
GR539525R1 |
May 05/1993 |
|
|
GB539525R4 |
Oct. 27/1999 |
|
|
GB539525R1 |
May 05/1993 |
|
|
FR539525R4 |
Oct. 27/1999 |
|
|
FR539525R1 |
May 05/1993 |
|
|
FI9200955W1 |
Jan. 23/1992 |
|
|
ES2139580T3 |
Feb. 16/2000 |
Vitaleteina y su Uso en el Cultivo Celular y en Terapia |
|
ES539525R4 |
Oct. 27/1999 |
|
|
ES539525R1 |
May 05/1993 |
|
|
EP9200955W1 |
Jan. 23/1992 |
|
|
EP539525B1 |
Oct. 27/1999 |
Vitaletheine and Use in Cell Culture and Therapy |
|
EP539525A4 |
Jan. 12/1994 |
Vitaletheine and Use in Cell Culture and Therapy |
|
EP539525A1 |
May 05/1993 |
Vitaletheine and Use in Cell Culture and Therapy |
|
DK9200955W1 |
Jan. 23/1992 |
|
|
DK539525R4 |
Oct. 27/1999 |
|
|
DK539525R1 |
May 05/1993 |
|
|
DE69131752C0 |
Dec. 02/1999 |
Vitalethin und Seine Anwendung in Zellkultur und Therapie |
|
DE539525R4 |
Oct. 27/1999 |
|
|
DE539525R1 |
May 05/1993 |
|
|
CH539525R4 |
Oct. 27/1999 |
|
|
CH539525R1 |
May 05/1993 |
|
|
CA2087884AA |
Jan. 11/1994 |
Vitaletheine and Use in Cell Culture and Therapy |
|
BR9200955W1 |
Jan. 23/1992 |
|
|
BG9200955W1 |
Jan. 23/1992 |
|
|
BE539525R4 |
Oct. 27/1999 |
|
|
BE539525R1 |
May 05/1993 |
|
|
BB9200955W1 |
Jan. 23/1992 |
|
|
AU9200955W1 |
Jan. 23/1992 |
|
|
AU8500891A1 |
Feb. 4/1992 |
Vitaletheine and Use in Cell Culture and Therapy |
|
AU655598B2 |
Jan. 05/1995 |
Vitaletheine and Use in Cell Culture and Therapy |
|
AT539525R4 |
Oct. 27/1999 |
|
|
AT539525R1 |
May 05/1993 |
|
|
AT186048E |
Nov. 15/1999 |
Vitalethin und Seine Anwendung in Zellkultur und Therapie |
IBM's former database indicates there also is a rather extensive family of patents that can be found by searching "Taub Floyd", many if not all of which appear to contain much the same compounds and utilities as the earlier authentic patents and applications properly naming Knight, Mann, and Scallen, or Knight and Scallen as inventors. It was admitted to the USPTO, and by both of UNM's so-called "expert witnesses" (who demonstrably couldn't follow chemical recipes for the authentic compounds) that the Taub, et al. patents and applications falsely claimed inventorships of the same compounds and uses discovered by Galen and Dr. Scallen, but no legal action for fraud has been taken, yet, against Taub and his accomplices by the courts of this country. Patents alleging Taub, et al., as inventors all seem to be based upon three provisional applications acknowledging the use of Galen’s and Dr. Scallen’s work......
US 60/075,966
US 60/085,474
US 60/005,366
......in which the vitaletheine modulators are described, for example, as carbonate esters of beta-alethine instead of "vitalethine", or as disulfides and other sulfur compounds with up to 3 amino acids per position in the structure, including modified peptides. Cysteamine and beta-alanine, both building blocks for beta-alethine and vitalethine, are among the contemplated amino acids and modified peptide components for most of these patents and applications alleging Taub, et al., as inventors. The District Court of New Mexico has declared that only Drs. Knight and Scallen are inventors of the vitaletheine modulators.
GALEN’S TECHNICAL REPORTS AND PRESENTATIONS
- Lovelace Continuing Medical Education Program (Channel 27)-October, 1997, Public access broadcast around New Mexico on Environmental Health, Nutrition, Immunotherapy.
- Channel 13, CBS Evening News-Presented nutritional information for better health.
- Channel 4, NBC Evening News-With PIRG, presented information and comments to Steve Schiff concerning environmental issues, and was interviewed afterwards about these concerns.
- Computer Expo-1996, Seminars on Publication of Scientific Information through the internet.
- New Mexico Environmental Health Conference-October 12-15, 1998, Presented a variety of environmental concerns that relate to and are independent of the vitaletheine modulators.
- Good Health, Good Life Conference-November 1 & 2, 1997-Organizer, and speaker for seminar providing continuing education credits for nurses.
- Good Health, Good Life Show (Channel 27)-Frequent guest on a variety of health issues.
- Facts about Pesticides & Safe Alternatives-Panelist in a discussion hosted by Northern New Mexico Community College, August 26, 1999
- American Society for Cell Biology-Two posters on beta-alethine and vitaletheine modulator technologies, November 15-19, 1992.
- American Association for Cancer Research Meeting-November 7-11, 1994, Poster presented in Taipei, Taiwan at the Conference on Modern Developments in Cancer Therapeutics.
- Source for Educational Empowerment and Community Development (SEED Open University)-1997 to present, Board Member, Secretary, Instructor.
- SEED THOUGHTS (Channel KHTL 920 AM)-August 21, 1998, June 12, 1998, Discussion on various health and social issues.
- University of Natural Medicine-1998 to present, Faculty and Advisor on Environmental Medicine, Nutrition, Immunotherapy, and Chemistry.
- Connections Radio Program (Channel KFSR 90.7)-Carly's and Diego's frequent guest speaker.
- HealthSouth Rehabilitation Hospital-Frequent guest speaker for their support groups including lymphedema, chronic fatigue, and fibromyalgia.
- United Silicone Survivors of the World-Frequent guest speaker on a variety of health issues for their support group.
- Sandia National Laboratories-Seminar on toxicology, nutrition, and immunotherapy.
- University of Arizona-Seminar on work in toxicology, nutrition, and immunotherapy.
- TPL, Inc.-Seminar on work in toxicology, nutrition, and immunotherapy.
- H.L. Snyder Memorial Research Foundation-Seminar on work in toxicology, nutrition, and immunotherapy.
- US Patent Office-Responses to office actions, declarations, and protests filed concerning the vitaletheine modulator patents and applications.
- Ann Wigmore Foundation-Frequent guest speaker on topics of toxicology, nutrition, and immunotherapy.
- Cell Robotics, Inc.-Seminar on toxicology, nutrition, and immunotherapy.
- Wild Oats Community Market-Seminar on toxicology, nutrition, and immunotherapy.
- University of Colorado-Seminar in Colorado Springs on toxicology, nutrition, and immunotherapy.
- University of Minnesota-Seminar on toxicology, nutrition, and immunotherapy.
- The Lovelace Institutes-Seminar on toxicology, nutrition, and immunotherapy.
- Pharmacia Adria-SP, Inc.-Seminar on toxicology, nutrition, and immunotherapy.
- Hauser Chemical (hereinafter "Hauser")-Brief seminar returning from AACR meeting.
- Texas Citrus Exchange-Seminar on toxicology, nutrition, and immunotherapy.
- Chronic Fatigue Support Group-Seminar on toxicology, nutrition, and immunotherapy.
- Meditrend-Seminar on toxicology, nutrition, and immunotherapy.
- The web site, http://www.VitaleTherapeutics.org.
- Intertribal Bison Council’s Denver Meeting-Presented nutritional advantages of bison over other meat sources, along with a rationale for why changes in the Native Americans’ diets have so decimated their health.
- First Nations’ Meeting in Albuquerque-Guest Attendee distributing over 400 copies of VitaleTherapeutics’ web site and the USDA databases on business-card-sized CDs during November, 2002, alone.
- Investment Bankers and Scandinavian
Clinical
Researchers-7 hour presentation January 12, 2003, investigating the
feasibility
of overseas trials to corroborate in human trials results observed in
veterinarian
models.
B. GALEN'S EDUCATION, EMPLOYMENT, AND HONORS:
Educational Highlights
Degree: Doctor of Philosophy in Chemistry
Institution: The University of Texas at Austin, Austin, TX, USA
Emphasis: Analytical, Organic, Physical and Bio Chemistry,
Endocrinology, Physiology, Drug Metabolism, Enzymology, Membrane
Biochemistry, Comparative Biochemistry, and Oligonucleotide
Biosynthesis Theory.
Degree: Bachelor of Science in Chemistry and Biology
Institution: Southwestern College, Winfield, KS, USA.
Emphasis: Physical, Inorganic, Organic, and Bio Chemistry,
Biology, Physics, Anatomy, and Mathematics.
Honors: Summa Cum Laude (3rd)
- Presidential Scholar
- Order of the Mound
- Teaching Assistant concomitant biochemistry course work.
- Scholastic Achievement Award from the American Institute of Chemists
- Associate Research Member, H.L. Snyder Memorial Research Foundation
Certification: Radioisotope Theory and Techniques, Oak Ridge Associated Universities
Galen's Employment Highlights
1994-Today
VitaleTherapeutics, Inc., Albuquerque, New Mexico, USA
Position: Founder, Director, President
Knight identified:
- new reasons for concern about chemicals widely used in our environment;
- a novel way of DEtoxing the body which is not only non-toxic, but probably capable of reversing and even eliminating some health problems and risks;
- nutritional factors with therapeutic potential in cancer, heart, and other immunological diseases, and delineated mechanisms for their action;
- new, logical approaches for treating AIDS and autoimmune disease;
- vitalethine bound to the monooxygenase purified from animal tissues;
- that cancer and heart disease should be prevented by optimizing this process;
- tumor-negating mechanisms for the vitaletheine modulators and for other therapeutic agents that appear to directly complement these immune regulators;
- that these approaches can be used therapeutically in veterinarian applications;
- that bison top round, and oat bran and
meal, are good sources of L-cystine;
- soy beans
and a variety of soy products reportedly have a good amount of
L-cysteine and L-cystine (USDA), but unfortunately, this is made
largely unavailable
by various soy
protease inhibitors like cystatins,
soyacystatin, Kunitz inhibitor, Bowman-Birk inhibitor, along
with extensive problems
created by extensive
processing in trying to get rid of these inhibitors, as well as toxins
like isoflavones
and metal
toxins from processing vats that likewise destroy or make much
of the L-cystine and L-cysteine unavailable
or less
nutritious;
- various nutritional supplements and herbs that diminish pain and inflammation without poisoning immunity;
- nutritional and herbal formulations being evaluated for commercial development;
- fundamental problems with fats in processed foods since i) hydrogenation of fats further depletes the limited supply of dietary omega-3 fatty acids whose ratios have ii) steadily declined relative to omega-6 fatty acids with a concomitant dramatic increase in “dis”ease;
- that he trans fatty acids resulting from heating and hydrogenation are NOT readily metabolized and tend to serve as substrate for artifactual epoxidation that is detrimental to processes controlled by vitalethine and its monooxygenase receptor.
1983-1994 University of New Mexico, Albuquerque, New Mexico, USA
Position: Inventor, Research Scientist
Knight identified:
- the vitaletheine modulators and new therapeutic uses, including those for ß-alethine;
- that vitaletheine modulators were probably present in biological extracts;
- ways of chemically synthesizing them;
- appropriate biological testing in animal models and cell culture, including revival of an abandoned immunological assay;
- biological assays that, once perfected, enabled the discovery, development, safety determinations, and FDA-approval of novel cholesterol-lowering drug, Lescol (®Sandoz Pharmaceuticals, now Novartis);
- indications that HMG-CoA reductase, and presumably cholesterol biosynthesis and ras oncogene expression (cancer) could be controlled with same process; and
- likely enzymes for the natural down-regulation of both cancer and heart disease.
1976-1982
The University of Texas at Austin, Texas, USA
Position: Graduate Student, Teaching Assistant
Knight identified:
- a physiologically relevant peroxide-independent thyroidal iodination activity as the probable true pathway for the biosynthesis of thyroid hormone;
- component parts of this system including thiol factors influencing activity;
- other conditions necessary to reconstitute the iodinating biological activity;
- suitable thiol substrates for the monooxygenase as a function of their pKa;
- phosphopantetheine as the probable biological precursor of these thiols.
1974-1976 H. L. Snyder Memorial Research Foundation /
Southwestern College, Winfield, Kansas, USA
Position: Undergraduate Student, Teaching Assistant, Research
Assistant
Knight used:
- affinity columns for purification of enzymes affected by carcinogens;
- chemical syntheses to prepare these affinity columns; and
- chemical carcinogens and enzymology to study cancer in laboratory animals.
II. MATERIALS REVIEWED:
Galen has reviewed all of the scientific literature mentioned above
including that documented in his patents, patent applications,
publications in scientific journals, and on VitaleTherapeutics’ website. He also has
reviewed extensive, related material in a variety of databases,
including MedLine, Hyperhealth, Mother Nature's Library, USDA
Nutritional databases, CAS-Online, and in an exhaustive search of the
chemical, biological, medical, and patent literature. Thus, Galen has
reviewed far more than the following specific documents and scientific
information, but includes only the following important literature for
the sake of brevity:
A. CARBAMIC/CARBAMINO/CARBONIMIDIC ACID TAUTOMERS AND CARBONIC
ESTERS OF Beta-ALETHINE
- Morrow, J. S., Keim, P., and Gurd, F. R. N. CO2 Adducts of certain amino acids, peptides, and sperm whale myoglobulin studied by carbon 13 and proton nuclear magnetic resonance. J. Biol. Chem., 249(23):7484-7494, 1974.
- Morrow, J. S., Keim, P., Visscher, R. B., Marshall, R. C., and Gurd, F. R. N. Interaction of 13CO2 and bicarbonate with human hemoglobulin preparations. Proc. Nat. Acad. Sci. USA., 70(5): 1414-1418, 1973.
- Yeable, P.L., Lochmüller, and C.H., Henkens, R.W. 13C nuclear magnetic resonance studies on the mechanism of action of carbonic anhydrase. Proc. Nat. Acad. Sci., USA., 72(2): 454-458, 1975.
- Patterson, A. and Ettinger, R. VII3. Nuclear magnetic resonance studies of the carbon-dioxide-water equilibrium. Z. Elektrochem., 64: 98-110. 1960.
- Ettinger, R., Blume, P., and Patterson, A. C13 chemical shifts in CO and CO2. J. Chem. Phys., 33: 1597-1598, 1960.
- Matwiyoff, N. A., and Needham, T. E. Carbon-13 NMR spectroscopy of red blood cell suspensions. Biochem. Biophys. Res. Com., 49(5): 1158-1164, 1972.
- Griffey, R. H., Scavini, M., and Eaton, R. P. Characterization of the carbino adducts of insulin. Biophys. J. 54: 295-300, 1988.
- 13C-NMR spectra of authentic beta-alethine, vitalethine, vitaletheine V4, and the sulfenate-linked benzyl dimer of the activated form of vitalethine.
- Provisional U.S. patent applications, 60/075,966 and 60/085,474, stating falsely that anti-melanoma effects of beta-alethine were reported in Cancer Research 54: 5623-5642, 1994.
- Provisional patent application, US 60/005,336, a resulting patent, US 6,007,819, and others that cover the beta-alethine and vitaletheine modulator compounds (carbonate esters of beta-alethine) and utilities, but allege Taub, et al., instead of Knight, et al., as inventors.
- Osake, A. Preparation of Isocyanates. Chem. Rev., 72: 457-496, 1972.
B. SULFONATE/SULFINATE/SUFENATE/SULFIDE/SULFHYDRYL/THIOL/DISULFIDE SPECTRA AND DATA
- All available data from Drs. Shield Wallace (PAI), Christopher Murray (Hauser Chemical), Floyd Taub (Dovetail Technologies), and Knight's, Mann's and Scallen's work;
- NMR and other data provided by Dr. Jan Dahmen;
- All mass spectra data on the authentic compounds supplied by Los Alamos National Laboratory and Brüker Instruments;
- All 93 scientific papers cited in the November 1, 1994, issue of Cancer Research (54: 5623-5635) on the vitaletheine modulators;
- All 38 scientific papers cited in the November 1, 1994, issue of Cancer Research (54: 5636-5642) on the biological activities of beta-alethine;
- Roeder, S. B. W., Master, B. S., and Stewart, C. J. 13C NMR of Coenzyme A and its constituent moieties. Physiol. Chem. & Phys., 7:115-123, 1975;
- Fieser, L.F., and Fieser, M. Reagents for organic synthesis, Vol. 1., pp. 296-310, especially p. 307, New York: John Wiley and Sons, Inc., 1967;
- Lowe, G. L. Oxidation of thiols and disulfides to sulfonic acids by dimethyl sulfoxide. J. Org. Chem., 41: 2061-2064, 1974;
- To the extent available for each
authentic and alleged reproduction of an authentic compound, the
following general categories of data and information:
a. Biochemical pathways for syntheses13C-NMR spectra before and after lyophilization
b. Recipes for chemical syntheses
c. Mass spectra before and after oxidizing at alkaline pH
d. Melting points with and without barium hydroxide traps
e. Thermal decomposition before and after neutralization with ZnO
f. Chemical derivatization before and after heating
g. Chemical derivatization before and after reduction
h. IR spectra before and after thermal decomposition
i. IR spectra before and after oxidation at alkaline pH
j. Mass spectra before and after reduction
i. 13C-NMR spectra before and after reduction
j. 1H-NMR spectra before and after reduction
k. 1H-NMR spectra before and after lyophilization - The HyperHealth database for chemical and nutritional insights, including identifying errors in chemical content.
III. OPINIONS EXPRESSED AND THE BASIS AND REASONS THEREFOR:
A. PROBLEMS OF OXIDATION IN SYNTHETIC ATTEMPTS OF OTHERS
1.
Mass Spectra and IR Evidence--discrepancies between authentic and
alleged attempts
Galen has pointed out time and again the
well-known
reaction of halines and dimethyl sulfoxide that often generate dimethyl
sulfide
as a byproduct. This information was compiled in Feiser and Feiser's
Organic Reactions (supra), as cited in the Cancer
Research article on the vitaletheine modulators (Knight, et al.), and it can
be inferred from well-known greater oxidative potential of mixed
halines.
Thus, haline contamination (chlorine and bromine) probably was
problematic
in the synthetic attempts of others using iodine.
Sulfenate-linked Benzyl Carbamate Dimer
Galen has since confirmed this problem encountered by others in an experiment in which the pH of the reaction to make the sulfenate-linked benzyl carbamate dimer was intentionally made alkaline, by overcompensating with base (calcium hydroxide to high pH) and then correcting back to the proper pH with hydrochloric acid, a mistake that someone unfamiliar with the sensitivity of this reaction to pH and haline contamination logically might make. The combination of high pH, even for a few minutes, and contamination with chloride ions from the hydrochloric acid solution resulted in a product contaminated with higher oxidation states for sulfur than has ever been determined for the authentic sulfenate-linked benzyl carbamate dimer. (Note that iodine is purer if one uses the crystals that form on the lid and sides of the glass container through sublimation instead of those contained in the bulk of a commercial product. Thus, purification of iodine through sublimation, prior to use, may provide distinct improvements in the chemical purity of the synthetic authentic compounds.)
The mass spectra of this artifact displaying higher oxidation states suggests that some of the structure of the original sulfenate-linked benzyl carbamate dimer remains intact, including a possible dimer composed of one sulfenic acid moiety (-S-O-) and one sulfinic acid moiety (-SO2-). As expected for artifacts having an additional oxygen atom, new mass ions were observed with molecular weights about 16 mass units higher than the bulk of the material, the majority apparently still being the authentic sulfenate-linked benzyl carbamate dimer. Confirming modest differences were also evident in the infrared (IR) absorption spectra of this oxidized preparation, showing a fine splitting of the single absorption band @~1200 cm-1 for the authentic compound. Note that more oxidation leads to more S-O and S=O absorption bands in this region of the IR spectra, as graphically illustrated in the overt and extensive artifactual oxidation observed in other's attempts (top two spectra) when compared to the authentic compound (bottom spectra).
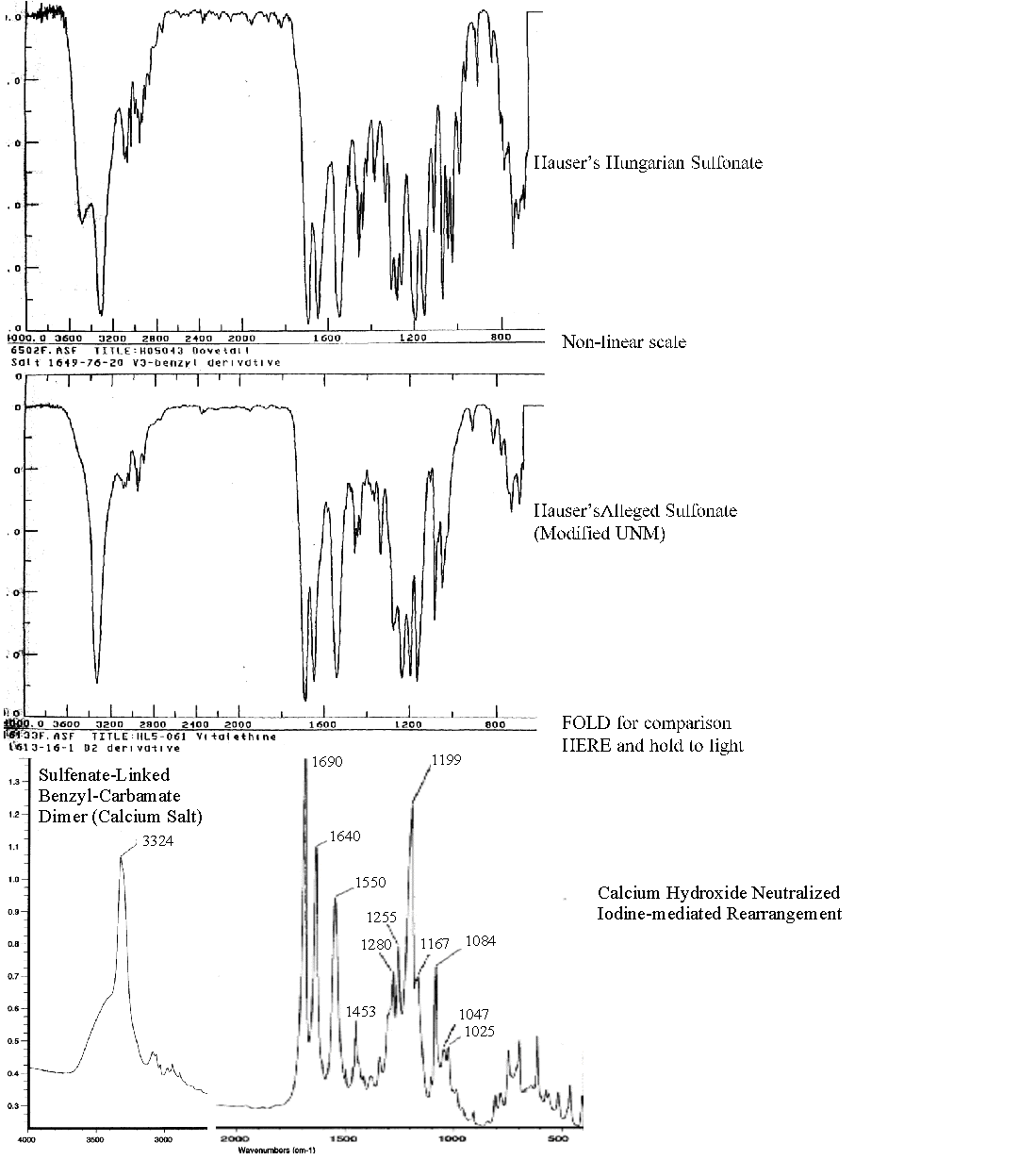
The mass spectra of others' attempt to make the sulfenate-linked benzyl carbamate dimer also indicates that this chemical company artifactually oxidized the sulfenate-linked benzyl carbamate dimer to higher oxidation states than the authentic compound, ones that may be linked through both sulfenate and sulfinate bonds (a "mixed linkage"), or by two sulfinate moieties. The calculated masses for a mixed, oxidative artifact (757.48) and for a dehydrated artifact of the sulfinate-linked benzyl derivative dimer (755.46) agree well with the masses actually observed by this company for their failed synthetic attempt. The artifacts so produced would lack the ability of the authentic compound to readily and spontaneously activate to biologically active sulfenate salts in biological systems. Diminished biological potency for products of these failed synthetic attempts, especially when compared to the consistent and potent biological activities of the authentic preparations, has been reported to Drs. Knight and Scallen by Dr. Paul Mann (unpublished results).
Although Galen's intentionally oxidized artifact of the sulfenate-linked benzyl carbamate dimer has a slight, possible indication for the calcium sulfonate @ 368.78-369.4 (calculated as 369.427), there is little evidence, if any, for this artifact in the mass spectra of the authentic calcium salt. In contrast to the authentic preparations, evidence for the sulfonate artifact is pronounced @ 392.94-394.89 in others' alleged attempts to make the authentic compound (calculated as 394.73 for the zinc salt).
Galen has chemically synthesized the pure zinc salt of the authentic sulfenate-linked benzyl carbamate dimer, finding it to have chemical properties, such as IR, very similar, if not identical, to the authentic calcium salt. Although an average mass ion of 742.48 would normally have been expected for the intact compound, the mass ion actually observed for the authentic zinc salt was @~570 (D2O) consistent with a partial, spontaneous breakdown of the sulfenate-linked benzyl carbamate derivative to a calculated mass ion of 569.96 in the high vacuum of the mass spectrometer. ZnO sublimes (evaporates) at room temperature, so it would be expected to be lost, in part, in much the same way that calcium is pulled from the sulfenate-linked benzyl carbamate dimer by calcium pumps within each mammalian cell. Thus, partial breakdown of the zinc salt was expected, as also predicted in the activation of the calcium salt in mammals. Either mineral-removing process facilitates at least partial activation of the authentic sulfenate-linked benzyl carbamate dimer to two molecules of vitaletheine's sulfenate (VSOH). Differences in stability between the zinc and calcium salts are also reflected in the higher melting point observed for the authentic calcium salt when compared to the authentic zinc salt.
Sulfenates (e.g., VSOH) are thought to be the quintessential biologically active forms of the vitaletheine modulators, the sulfenate-linked benzyl carbamate dimer, as pronutrient thereof, being affected less by the presence of the dead cancer cells than thiol forms of the vitaletheine modulators, including vitalethine that must cycle through a reduced form (aliases thiol, sulfide, or sulfhydryl) before being oxidized by the monooxygenase to sulfenic acids. Thiol forms (such as vitaletheine, VSH) are known to react with, and are probably inactivated by, the lipid epoxides formed in dead, cancer cell membranes ("ghosts"). In essence, this trapping of the reduced forms of vitaletheine modulators probably explains the increased cancer and heart disease risks associated with the dietary consumption of rancid or oxidized unsaturated fats such as those found in Fat Fried Fast Food (Quad Fs for "F"ailing health). This concept also explains the widely recognized health benefits of dietary antioxidants in countering tendencies of unsaturated fat and cancer cell ghosts to go rancid, to poison immunity, and to increase the risk of cancer, and its recurrence, and heart disease.
When the partially oxidized preparation of the authentic sulfenate-linked benzyl carbamate dimer was reduced in the MALDI mass spectrometer with iodide, the amount of overly-oxidized forms detected before and after reduction did not change, but there was a radical loss of higher molecular weight ions, those associated with the authentic sulfenate-linked benzyl carbamate dimer, with a concomitant increase in smaller masses more characteristic of vitaletheine and polymers, thereof. This is as expected, since the authentic sulfenate-linked benzyl carbamate dimer theoretically is capable of being reduced to these forms under these conditions, while the sulfonate structure alleged by others is not. Evidence for cyclization and polymerization of vitaletheine is also consistent with the difficulties encountered, below, when trying to derivatize vitaletheine, the reduced form (sic., thiol, sulfide, and sulfhydryl) of the disulfide, vitalethine.
2. NMR-discrepancies between authentic and alleged attempts
Galen also has pointed out, time and again, problems caused by over-reliance upon 13C-NMR in interpreting results of studies on this family of compounds, a potential problem also documented by Roeder, et al. This warning appears to be unheeded by others attempting unsuccessfully to synthesize the authentic compounds. Consequently, a detailed comparison of NMR spectra for the authentic compounds with those from the synthetic attempts of others is warranted.
Sulfenate-linked Benzyl Carbamate Dimer
According to proton NMR spectra supplied, products of others' alleged attempts to make the authentic sulfenate-linked benzyl carbamate dimer are not consistent with the authentic compound, nor are their attempts using a modified "authentic method" even consistent with their own attempts using alternative synthetic approaches to the sulfonate artifacts. Careful comparison of the proton NMR spectra indicate that the chemical shifts for methylene carbons immediately adjacent to the sulfur move progressively upfield (smaller numbers) as the products are oxidized from the authentic compound. This is evident in others' attempts allegedly using an admitted alteration of the "authentic method", especially when compared to attempts using a different sythnthetic approach to the alleged sulfonate compounds, essentially methods not even designed to yield the authentic compounds. These subtle discrepancies in the NMR results between the authentic compounds, and others' attempts to reproduce same, are consistent with the discrepancies noted in the IR spectra as the latter compounds are more extensively oxidized, thereby confirming again the discrepancies noted, supra, using both IR and mass spectrometry.
Lyophilization (freeze drying) of the sample is a common disastrous departure from the authentic procedures in the synthetic attempts of others. The authentic compounds were never lyophilized, since this procedure has been shown to radically alter the product chemically. Problems with such treatments were noted by Galen long ago, having since been expanded upon on this website. Furthermore, there is a wealth of scientific literature, supra, making it clear that the bonds in the carbamic/carbonimidic acid tautomers of vitaletheine V4 and vitalethine should be labile at low partial pressures of CO2. Considering the very low partial pressures of CO2 maintained within the MALDI mass spectrometer by turbo pumps, it is no small wonder that ANY indications for the carbamic/carbonimidic acid moieties were detected in the MALDI mass spectra of authentic vitalethine and vitaletheine V4, and especially in the mass spectra of authentic beta-alethine. Since no heterogeneity is observed in the NMR of these samples, it is likely that the heterogeneity observed in these analyses result from the high vacuum and the ultraviolet laser blast used to volatilize the sample for these mass spectrometry determinations, especially the laser-induced polymerizations or adducts of the original compounds similar to those polymerizations observed with UV light in the chemical synthesis of vitaletheine V4.
Vitaletheine V4
Discrepancies in proton NMR spectra, between the authentic preparations and the alleged synthetic attempts by others, are even more pronounced for vitaletheine V4 than for the sulfenate-linked benzyl carbamate dimer. As before, the NMR shifts for the methylenes adjacent to the sulfur are not the same for i) others' synthetic attempts allegedly using authentic procedures and ii) results of other synthetic approaches to the alleged sulfonates. Secondly, others' lyophilized preparations, allegedly of vitaletheine V4, had dramatic downfield shifts (higher values) in all of its methylenes compared to an authentic sample, possibly due to the combined effects of oxidation and lyophilization. There does appear to be a consistent and pronounced chemical effect of dehydration and/or decarboxylation upon authentic vitaletheine V4, brought about by the simple act of freeze-drying. Others' preparations, allegedly produced using the authentic method, suffer these downfield shifts relative to the authentic compounds, indicating that attempts by others to synthesize authentic compounds resulted in artifacts due to the combined effects of oxidation, dehydration, and/or decarboxylation in the lyophilization process.
Methylenes adjacent to the sulfur in authentic vitaletheine V4 are upfield (smaller numbers in the authentic compound) relative to those in others' synthetic attempts), whether using another synthetic approach to the alleged sulfonates or an admittedly altered version of the authentic method. As with attempts at making the authentic sulfenate-linked benzyl carbamate dimer, these discrepancies, between the spectra of authentic vitaletheine V4 and the results of others’ alleged attempts to make the authentic compound, indicate that higher oxidation states and perhaps even sulfonates result when one departs from the authentic methods. These radical departures from the authentic protocols, such as inclusion of an additional step using hydrobromic acid and lyophilization, indicate that there probably were NO serious attempts by these others to reproduce the authentic work, serious departures from the recipes for the authentic compounds being most obvious by those falsely claiming inventorships of the same compounds and uses discovered by Galen and Dr. Scallen.
Although differences in chemical shifts are noted in the proton NMR of the authentic compounds, when compared to the synthetic attempts of others, the most startling and telling differences in the products of these two synthetic approaches for vitaletheine V4 are in the proton NMR coupling constants. Lyophilized authentic vitaletheine V4 has a coupling constant much larger than the lyophilized product of either alleged approach by others. A large coupling constant indicates that the authentic compound has a highly constrained ring system involving the sulfur atoms, instead of the straight-chained and uncontrained sulfonates alleged by others. Sulfur atoms, because of their sheer bulk, typically occupy a staggered, right-angled conformation, which tends to minimize coupling constants. Any departure from this staggered conformation to a more eclipsed conformation, such as those produced by steric forces in the constrained authentic tetramer, tends to increase coupling constants. Thus, the proton NMR-spectra of authentic vitaletheine V4, when lyophilized, appears to contain direct evidence for a more eclipsed conformation, compared to the unconstrained, straight-chained "simpler" structures alleged by others. This large coupling constant, then, supports the structure of the authentic compound containing rings, as has been proposed for the tetramer, that produces steric constraint under certain dehydrating circumstances, such as when lyophilized. It is very interesting that the authentic vitaletheine V4 does not exhibit large coupling constants until it is lyophilized. This suggests that the authentic tetramer is not as constrained before lyophilization as after, and is, therefore, much more stable before lyophilization than others, falsely alleging inventorship, would like for us to believe.
Since this large coupling constant was not observed in the proton NMR spectra of others' lyophilized attempts to synthesize the authentic compound, it is highly unlikely that others have made stable, authentic preparations of vitaletheine V4. A possible exception is one preparation made that decomposed with the evolution of gas (CO2?) soon after the proton-NMR was acquired. Proton NMR of this one unstable preparation did indicate that the terminal amine was still bound to something (CO2?), since it displayed a triplet, much like that of the CBZ-blocked starting material, instead of a higher multiplet or broad peak that would be expected, and that has indeed been observed, when the carbon dioxide has been removed from the amine. As described above, lyophilization is one process through which this artifactual dehydration or decarboxylation can be produced. Thermal decomposition of the carbamic/carbonimidic tautomers is another way of removing the CO2 from the carbamic and carbonimidic moieties.
B. NMR AND OTHER EVIDENCE FOR THE CARBONIMIDIC TAUTOMER
1.
Carbon NMR Evidence
The proton and carbon NMR for the authentic vitalethine, beta-alethine, and vitaletheine V4 preparations are all consistent with the structures reported in US 5,370,868 and in the back-to-back November 1, 1994, Cancer Research articles.
It is difficult to get the carbon spectra of the carbonimidic tautomer, since one must have high concentrations of sample when the compound has only the low, natural abundance of 13C. Thus, a sample of at least 100 mg/ml of the vitaletheine modulator is probably needed to see the natural abundance of 13C in the NMR of carbamic/carbonimidic tautomers. This is made obvious by the works of others using such adducts of carbon dioxide with the amino groups of hemoglobin. Without isotope-enriched carbon dioxide one would probably have to have a hemoglobin concentration of 4 grams/ml or more to observe the carbonimidic tautomer. Matwiyoff and Needham (supra) apparently were barely able to observe what appears to be a carbonimidic tautomer in hemoglobin using 13C-enriched carbon dioxide and 0.13 grams of hemoglobin per milliliter. Even if one could get 4 grams of hemoglobin into one ml of solution, under this circumstance, the natural abundance of the other carbons in the hemoglobin would probably swamp out the signal for the carbonimidic acid. Consequently, it is rather fortuitous that Galen was able to demonstrate the NMR signal for the carbonimidic tautomer of vitalethine, at all, in authentic preparations of vitalethine and vitaletheine V4, both preparations having only the natural abundance of 13C. Galen was able to do so only by using high concentrations of the synthetic compounds in the 13C-NMR analyses. Note that the chemical shift for the carbonimidic tautomer of these two vitaletheine modulators are in a range expected, by extrapolation of the additivity rule, and according to the chemical shifts observed by Matwiyoff and Needham for the probable carbonimidic tautomer of hemoglobin (~114-115 ppm relative to TMS).
Vitaletheine V 4
Although the chemical shift for the carbonimidic tautomer of authentic vitalethine is NOT in the range expected for isocyanates (as alleged by others) nor even in the range expected for solubilized carbon dioxide, it is important to note that authentic vitaletheine V4 after lyophilization does produce a chemical shift in the range expected for isocyanates. It is conceivable that lyophilization could dehydrate the original carbamic/carbonimidic tautomers into isocyanate moieties. Furthermore, ionization is known to shift carboxylic acids downfield relative to protonated acids, so it is quite possible that pulling ZnO out of authentic vitaletheine V4 during lyophilization facilitates ionization and/or dehydration of the carbonimidic tautomer to a more downfield position than would be expected for the protonated, carbonimidate tautomer. Dissolved CO2 is a somewhat less likely explanation for this shift in the lyophilized vitaletheine V4, since the shift observed is upfield from that expected for CO2 and since the presence of any dissolved CO2 also should have generated a strong bicarbonate shift at about 161 ppm downfield from TMS. The latter was not observed, so a dissolved CO2 explanation for the observed peak is highly unlikely. Thus, lyophilization conceivably can increase steric constraints, ionize, and dehydrate the carbamic/carbonimidic tautomers of the authentic compound into isocyanate moieties, and cause other problematic dehydrations, decarboxylations, and rearrangements.
2.
Biochemical Evidence
Galen has discussed, time and again, the evidence in the literature for the carbonimidic and carbamic acid structures being stable in aqueous solution. There are 11 scientists who published peer-reviewed papers in scientific journals on this subject who have lived in the Albuquerque area, alone. Indeed, it could be argued that carbamic and carbonimidic acid tautomers are the basis for all plant and animal life as we know it, since i) the incorporation of carbon dioxide into sugars in photosynthesis and ii) the transportation of the waste product, carbon dioxide, away from the combustion of these sugars by animals cells, both depend upon these carbamic/carbonimidic acid complexes between the amino groups of proteins (in plants and animals) and carbon dioxide.
Vitalethine
Vitalethine is estimated to be between 100 million and 3 billion times more potent biologically than the authentic preparations of beta-alethine. Though postulated long ago, MALDI mass spectrometry produced the first evidence that authentic preparations of beta-alethine may contain traces of vitalethine. Carbonic acid esters of beta-alethine (vitalethine) can form, if through no other mechanism, by reaction of the hydrated free amines of absolutely pure beta-alethine with atmospheric carbon dioxide. Thus, any solution of beta-alethine can be considered to contain some vitalethine as a function of the partial pressure of carbon dioxide, the pH, and the time with which the beta-alethine is allowed to equilibrate with a carbon dioxide source, such as the bicarbonate buffer in the body fluids of animals. A carbonic acid ester of an amine is just another way of saying a carbamic (or carbonimidic acid tautomer), like the very moieties critical to the potency of authentic vitalethine.
This brings us to a logical inconsistency in provisional
applications
US 60/075,966 and US 60/085,474 falsely alleging Taub, et al., as inventors of the
beta-alethine and vitaletheine modulator technologies. These
provisional
applications claim that beta-alethine, when treated in a manner
allegedly
designed to remove counterproductive materials, results in a new
product,
BetathineTM. However, instead of increasing potency relative
to authentic beta-alethine preparations, as would be expected when
removing
the alleged counterproductive materials, there appears to be a
pronounced
decrease in the potency of BetathineTM relative to authentic
beta-alethine. The provisional applications report biological potencies
for BetathineTM that are probably tens of thousands of times
less
than the authentic preparations of beta-alethine. Thus, the method of
producing
BetathineTM in these provisional applications by Taub, et al.,
appears to remove a therapeutic component from beta-alethine, instead
of
or along with, the removal of any alleged counterproductive component.
The alleged presence of a counterproductive component in either authentic beta-alethine or authentive vitalethine is NOT supported by the proton or carbon NMR analyses of these pristine materials. In addition, at optimal therapeutic doses of only 100 fg/kg, any alleged contamination of vitalethine would have to be far more potent than vitalethine, itself, or face dilution beyond any possible therapeutic significance. Thus, the allegations of Taub, et al., of contaminations in either authentic preparation appear to be completely irrational, unfounded, and unwarranted.
An probable explanation for observed differences in these potencies is that the mass spectra of Taub, et al.'s alleged preparations of beta-alethine are obviously and admittedly contaminated by what appears to be a mixed disulfide of beta-aletheine and cysteamine for some masses and brominated derivatives for others, either of which could inhibit the biological activities of authentic beta-alethine or vitalethine, in much the same way that cysteamine is known to inhibit iodination reactions in the thyroid at higher concentrations than encountered biologically.
The potency of authentic beta-alethine also can be diminished to that of BetathineTM or Beta-LTTM by taking steps to remove traces of the more potent therapeutic agent, vitalethine, from the authentic preparation of beta-alethine (See mass spectra, supra). It is unlikely that vitalethine will ever be completely removed from beta-alethine in therapeutic applications for several reasons. Apparent efforts in the provisional applications (supra), to remove traces of vitalethine from beta-alethine, theoretically, can be reversed almost immediately by mere exposure of these depleted preparations to any source of carbon dioxide, such as the atmosphere or the bicarbonate buffering found in animal body fluids and tissues. It also has been alleged that beta-alethine produces a TH-1 response, instead of the TH-2 response to vitalethine observed in the mAIDS model by Drs. Knight, Watson, Wang, and others. Unfortunately, this report of Taub, et al., misrepresents results obtained by Knight, et al., as reported in back-to-back papers in Cancer Research, making other information provided and conclusions drawn by Taub, et al., suspect. For example, it remains to be determined if this effect of BetathineTM/Beta-LTTM is independent of the biological activities of vitalethine or if BetathineTM/Beta-LTTM, contaminated with a mixed disulfide of cysteamine and beta-alethine, produces the reported TH-1 response simply by inhibiting vitalethine's demonstrated TH-2 response. It is possible, as well, that beta-aletheine in its substantially pure form is reduced in the cell to become a non-specific lipid epoxide scavenger, that neutralizes lipid epoxides normally reacting with and inactivating the more potent endogenously produced vitaletheine. Dead cancer cells, presumably because their ruptured membranes contain lipid epoxides, have been shown to block therapeutic responses of vitalethine in the Cloudman S-91 melanoma model. This mechanism for beta-alethine's biological activities also would readily explain the radioprotective effects of high doses of authentic beta-alethine observed long ago in a collaboration of Galen and Dr. Scallen with the Armed Services Radiobiological Research Facility.
The conclusions of Taub, et al., are also made suspect by their use of the wrong solvent in the synthetic procedures for beta-alethine and ultimately vitalethine. The artifactual use of acetone, instead of acetonitrile, is suspected of producing a Schiff base artifact of vitalethine, capable of even mimicking the tautomerism of the carbamic/carbonimidic moiety of vitalethine. Chemical analogues of natural compounds, such as this Schiff base analogue of vitalethine, are known to inhibit the desirable activities of the natural substances. For example, competitive inhibitors of pantothenic acid, a building block of the vitaletheine modulators, have been known for some time to completely block humoral (sic., antibody) responses to vaccines.
Vitalethine dramatically stabilizes cells in culture at very low concentrations (~attograms/ml) compared to the effective concentrations of authentic beta-alethine (~nanograms/ml). Protection of vitalethine, and especially vitaletheine, by antioxidants, and by free-radical and epoxide scavengers, appears to be absolutely critical for a long and healthy life. Furthermore, supporting the monooxygenase receptor for vitaletheine helps to ensure that most of the vitaletheine is kept in more oxidized forms, its sulfenic acid (VSOH) or vitalethine (VSSV), that are resistant to reaction with epoxides. The induction of epoxide hydrolase by sulforaphane from the Cruciferous family of plants (such as broccoli sprouts) is another example of an epoxide scavenging mechanism that is thought to have profound health benefits, probably through the protection of vitaletheine produced naturally by the body.
Problems resulting from the use of such high doses of BetathineTM/Beta-LTTM (1-100 mg/kg) cannot be ignored. If BetathineTM/Beta-LTTM is an inhibitory foil of vitalethine, its therapeutic use in pure form (devoid of vitalethine) may be limited to inhibiting immunoproliferative, TH-2 directed cancers, and cancers resulting from TH-1 deficiencies. Indeed, the authentic preparation of beta-alethine was counterproductive in the not-quite-syngeneic melanoma model, whereas TH-2 directed, immunostimulating authentic vitalethine produced ~50% cure rates in this same model (US 5,370,868). Unlike authentic beta-alethine, that was 100% effective in a mouse myeloma model (NS-1 myeloma), the grossly-contaminated BetathineTM made by Taub, et al., was not able to stop the progression of cancer in 5 of 8 B-cell lymphoma human patients. These concerns are amplified by the life-threatening agranulocytosis caused by levamisole, a structural analogue of beta-alethine, and of the vitaletheine modulators, identified in the November 1, 1994, issue of Cancer Research. For these reasons, and the striking differences in potency and purity between authentic beta-alethine and BetathineTM/Beta-LTTM, BetathineTM/Beta-LTTM at the very least should have been shown to be effective and safe in a syngeneic cancer model, say the S-91 melanoma model, before it could be ethically advocated for clinical testing in human cancer.
For example, dramatic differences in the biological potencies for authentic beta-alethine and for the authentic vitaletheine modulators were observed in i) hematopoietic assays that measure the ability of progenitor cells to produce red blood cells, ii) in hemolytic plaque assays that measure antibody production, and iii) in various tumor models, such as not-quite-syngeneic melanoma and syngeneic myeloma mouse cancers. In all cases, even when the experiments were blinded, comparisons of the authentic preparations indicated that authentic vitalethine was about 100 million to 3 billion times more potent than authentic beta-alethine. These consistent differences in biological potencies in many different laboratories and assays simply cannot be ignored. Authentic beta-alethine and vitalethine obviously are chemically different, and these differences are confirmed resoundingly by the observed dramatic differences in their biological potencies. The differences between authentic vitalethine and BetathineTM/Beta-LTTM are even more pronounced, BetathineTM/Beta-LTTM reportedly being about 10 trillion times less potent than authentic vitalethine in biological assays. Aside from the obvious differences in chemical purity, side-by-side comparisons of the various synthetic preparations are needed in the same biological model to determine the relative potencies of the authentic vitalethine and beta-alethine and others' alleged and obviously contaminated preparations.
3.
Decompositions & Chemical Derivatizations
Derivatizing vitaletheine, unlike beta-aletheine and cysteamine, is difficult with sulfhydryl (thiol or sulfide) reagents such as monobromobimane. After many attempts under a variety of conditions, the following conclusions were drawn: vitalethine appears to either i) produce a thiol derivative that cannot be separated under conditions that all other thiol derivatives are, or ii) the reduced form, vitaletheine, cyclizes or polymerizes, probably via nucleophilic attack on the carbonyl of its amide linkages, immediately upon formation, thereby making the sulfhydryl moiety unavailable for derivatization. Thus, derivatives other than those for thiols were explored to prove the structure of vitalethine.
It was determined that free amines tend to destabilize carbamic acid structures, since aqueous ammonium carbamate decomposes at a lower temperature than aqueous solutions of other salts of this free carbamic acid, such as zinc carbamate. Thus, it was postulated that the heating of vitalethine might cause it to decompose and expose some free amine, thereby facilitating even further decomposition of the carbamic and carbonimidic acid tautomers and allowing the derivatization of amines, so liberated, with dabsyl chloride. When both are neutralized with ZnO, thermal decomposition of vitalethine and beta-alethine to a common end product can be demonstrated, by the disappearance of differences between them in their IR spectra after heating both to their melting points. A gas (CO2?), evolving from vitalethine (or vitaletheine V4) upon heating, can be trapped with aqueous solutions of barium hydroxide, probably as the precipitated and insoluble barium carbonate, an observation that confirms decompositions of their carbamate moieties.
Unheated, vitalethine can be derivatized
with dabsyl chloride only to a limited extent, the product
exhibiting little fluorescence and eluting early in a region more
typically associate with anionic (R-COOH containing) compounds than
with derivatives of beta-alethine (R-NH3+
containing). Dabsylation of heated vitalethine produces a derivative
that is more characteristic of those adducts obtained with cystamine
and beta-alethine, eluting late in the chromatogram. Thus, heating
enabled a more thorough and more intensely fluorescent derivatization
of vitalethine, one similar but not identical to the dabsylamide of
heated beta-alethine. At the same time, the derivatized product of
heated vitalethine elutes between those times
expected for derivatives produced from pure cystamine and from pure
beta-alethine.
One probable explanation for this is that the primary derivatized product of thermally-decomposed vitalethine could be a mixed disulfide of cysteamine and beta-aletheine. The byproduct of such a heterologous cleavage would be beta-alanine, an amino acid that should produce another anionic, early-eluting dabsyl amide. An intensely fluorescent peak, in fact, was observed eluting early in the region expected for such an anionic dabsyl amide. A mixed disulfide of beta-aletheine and cysteamine, produced from both vitalethine and beta-alethine upon heating, also would help to explain why the IR spectra of both heated vitalethine and heated beta-alethine are similar, but different from the spectra of unheated beta-alethine. It is not known at this time if the carbonate esters of beta-alethine (vitalethine) are in some catalytic way more effective at producing heterolytic cleavage of central amides upon heating than are the free amines of beta-alethine. Differences in the hydrochloric acid content of the beta-alethine before and after heating, obviously, could account for some of these observed differences in its IR spectra, but this should have been minimized by the neutralization of the beta-alethine preparation with ZnO prior to heating.
Other alternate explanations for the observed elution profiles include the possibility that anionic derivatives, conceivably more abundant in the heated vitalethine preparation than in the heated beta-alethine preparation, could contribute to an early elution of the dabsyl amides of more beta-alethine-like derivatives. It is interesting that a mixed disulfide of beta-aletheine and cysteamine appears to be contaminating others' synthetic attempts to make pure beta-alethine, as evidenced by their own mass spectra. Such artifacts were not observed in authentic preparations until treated in a manner expected to cause decompositions and rearrangements.
4.
Comparisons of Elemental Analyses
The elemental analyses of the authentic compounds were reported in authentic patents such as US 5,370,868. A comparison with the alleged sulfonates, taking into account evidence for ZnO loss from vitaletheine V4 and hydration of the sulfenate-linked benzyl carbamate dimer, was presented long ago by Galen. Although carbonate esters of amino-alkyl-sulfonates were contemplated in the original authentic patent applications, elemental analyses of the authentic exemplary compounds are inconsistent with the alleged sulfonate structure. Thus, the authentic exemplary compounds simply are not sulfonates, as falsely alleged by others, and especially are NOT amino-alkyl-sulfonates NOR are they N-carbobenzoxyl derivatives of same. That does not mean that the originally contemplated sulfonates are necessarily devoid of all biological activity, nor does it mean that the true inventors now feel the sulfonates have no value, it merely means that these compounds were contemplated but were not among those chosen to be exemplary compounds for the authentic patents and patent applications, a choice apparently quite justified by the striking disparity in the biological potencies of the authentic compounds compared to the sulfonates produced by others, as reported by Dr. Paul Mann to Galen and Dr. Scallen.
An elemental analysis reported by another for his attempt at making
the sulfenate-linked benzyl carbamate dimer was inconsistent with both,
the alleged sulfonate structure, and the quite accurate elemental
analysis for the authentic sulfenate-linked benzyl carbamate dimer.
Since Galen encountered a similar problem with an elemental analysis
for authentic vitalethine, a
commercial elemental analysis consistent with neither the vitalethine
nor
the beta-alethine structures, part of the inconsistencies in these
analyses may have resulted from a mishandling of these compounds by
commercial analytical laboratories. These problems were not encountered
with the elemental analyses performed by Ruby Ju, who took extra
precautions to ensure that the authentic samples were not inadvertently
decomposed, dehydrated, polymerized, or decarboxylated. Ms. Ju’s
results were as expected for the authentic exemplary compounds,
as reported in US 5,370,868, and in other patents and applications that
have not been corrupted by UNM and its now defunct licencee, Floyd
Taub, and his accomplices.
GO TO:
| Home | Overview | People | Journal | Nutrition |
| Environment |
|
WWW Links | Outline | e-mail us |